Why Do We Design Primers?
Primers are short (usually 18-27 bp), single-stranded DNA sequences that are complementary to the target DNA sequence. They are essential components in the Polymerase Chain Reaction (PCR) process, which amplifies a specific fragment of DNA. The main reasons why we design primers for PCR are:
1.Specificity: Primers ensure that only the region of interest is amplified and not other regions with similar sequences. Specific primers improve the accuracy of PCR and reduce false-positive results.
2.Efficiency: Primers must be designed to bind to the target DNA sequence with high affinity and stability, allowing efficient amplification of the desired product.
3.Length: The length of primers should be long enough to provide specificity and short enough to avoid non-specific binding.
4.Melting temperature (Tm): Primers should have a melting temperature (Tm) within a narrow range for efficient annealing during PCR.
5.Avoiding secondary structures: Primers should not form secondary structures like hairpins or loops that could interfere with PCR amplification.
Steps
- Grab your sequence region from NCBI (Variation/Gene Viewer), Ensemble, or UCSC genome viewer.
- Design primers with Primer3Plus either using the CLI version or the web interface.
- Analyse specificity with BLAST-Nucleotide (fwd primer).
- Check the dimerization on the PCR stats website.
- Primer3 protocols for primer-BLAST:
- Self complementarity < 8.00
- Self 3’ complementarity < 5.00
If you’re designing primers for sequencing (sanger or else), remember to always be at least 100 bp away from your target region.
Length
The length of a primer can affect its specificity, annealing temperature, and amplification efficiency in PCR. Generally, the length of a primer is chosen based on the target DNA sequence and the specific PCR reaction conditions.
If a primer is too short, it may anneal to non-target sequences in the template DNA, leading to non-specific amplification. Conversely, if a primer is too long, it may anneal less efficiently to the target DNA sequence, leading to reduced amplification efficiency. Therefore, an optimal primer length that balances specificity and efficiency is typically chosen.
The optimal primer length can vary depending on the specific PCR reaction conditions, such as the annealing temperature, salt concentration, and buffer composition. As a general rule of thumb, primers in the range of 18-24 nucleotides are commonly used in PCR.
In addition to specificity and amplification efficiency, the length of a primer can also affect the melting temperature (Tm) of the primer, which is the temperature at which the primer anneals to the template DNA. Generally, longer primers have higher Tm values than shorter primers, all other factors being equal. This is because longer primers have a higher likelihood of forming stable secondary structures, such as hairpins, that can increase the Tm value.
Long primers have a higher likelihood of forming secondary structures, such as hairpins, which can reduce the efficiency of primer annealing to the target DNA sequence. These secondary structures can also compete with the formation of the desired primer-template duplex, leading to reduced specificity and efficiency of the PCR amplification.
Melting Temperature
Tm, or melting temperature, is the temperature at which half of the DNA strands in a double-stranded DNA molecule are dissociated into single strands. Tm is a critical parameter in PCR (polymerase chain reaction) and other DNA-based techniques, as it determines the optimal temperature for primer annealing and template DNA denaturation.
The Tm value of a primer is influenced by several factors, including the length and nucleotide composition of the primer sequence, the salt concentration of the reaction, and the presence of other PCR components such as Taq polymerase, dNTPs, and PCR buffer. Generally, primers with higher GC content have higher Tm values, as GC base pairs have three hydrogen bonds, while AT base pairs have two hydrogen bonds.
In PCR, the annealing temperature is typically set slightly below the Tm of the primers to promote specific primer annealing to the template DNA. If the annealing temperature is too low, non-specific annealing can occur, leading to non-specific amplification. If the annealing temperature is too high, the primers may not anneal efficiently, leading to reduced amplification efficiency. Therefore, determining the appropriate annealing temperature is a critical step in PCR optimization, and it often involves testing a range of temperatures to determine the optimal annealing temperature for the specific primer-template combination and reaction conditions.
GC-Content
GC-content refers to the proportion of guanine (G) and cytosine (C) nucleotides in a DNA molecule, expressed as a percentage of the total nucleotides in the molecule. The GC-content is an important parameter in many molecular biology techniques, as it can affect the stability of the DNA molecule, as well as the efficiency and specificity of PCR amplification. This formula shows how to calculate the GC conent of a sequence:
$$GC_{\%} = \frac{G + C}{A + G + C + T} \times 100$$
To calculate the GC-content of a DNA sequence, you can follow these steps:
- Count the number of G and C nucleotides in the sequence.
- Count the total number of nucleotides in the sequence.
- Divide the total number of G and C nucleotides by the total number of nucleotides and multiply by 100 to get the GC-content as a percentage.
For example, consider the DNA sequence “ATCGATCGTAGCTAGCTAGC”. To calculate the GC-content:
- Count the number of G and C nucleotides: there are 6 G nucleotides and 5 C nucleotides, for a total of 11 GC nucleotides.
- Count the total number of nucleotides: there are 20 nucleotides in the sequence.
- Divide the total number of GC nucleotides by the total number of nucleotides and multiply by 100: (11/20) x 100 = 55%.
Therefore, the GC-content of the sequence “ATCGATCGTAGCTAGCTAGC” is 55%.
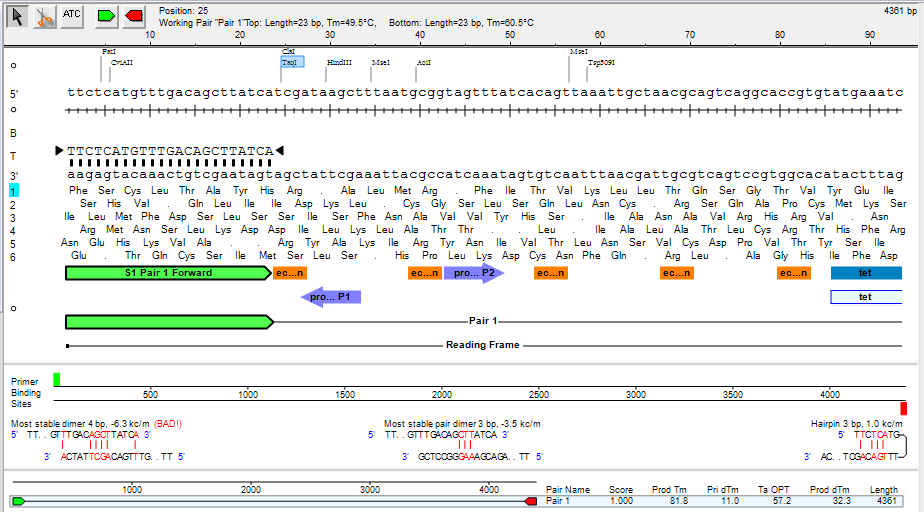
Dimerization
In general, the optimal \( \Delta G \) values for primer’s hairpin and self-complementarity (self-dimer) formations depend on the specific application and experimental conditions.
For PCR (polymerase chain reaction), the \( \Delta G \) value for the hairpin formation should be less than -3 kcal/mol to ensure efficient amplification. Similarly, for self-dimer formation, the \( \Delta G \) value should be less than -5 kcal/mol.
However, for other applications such as hybridization probes, the optimal \( \Delta G \) values may differ depending on the specific assay requirements. It’s important to consider the melting temperature (Tm) of the primer/probe in addition to \( \Delta G \) values to ensure specific and efficient binding.
In general, there are various online tools available to predict the \( \Delta G \) values for primer hairpins and self-dimer formations based on sequence information.
When designing primers for PCR (polymerase chain reaction), it is important to consider the possibility of hairpin, self-dimer, and hetero-dimer formations. These are secondary structures that can form when the primer sequences anneal to each other, which can lead to inefficient PCR or non-specific amplification.
Hairpin formation: Hairpin formation occurs when a single-stranded DNA molecule folds back on itself and forms a double-stranded stem-loop structure. This can happen when a section of the primer sequence is complementary to another section further down the same primer sequence. Hairpin formation can prevent the primer from annealing properly to the target DNA template and lead to reduced amplification efficiency.
Self-dimer formation: Self-dimer formation occurs when two copies of the same primer anneal to each other, forming a double-stranded structure. This can happen when the primer sequence contains regions that are complementary to each other, resulting in the formation of a stable duplex structure. Self-dimer formation can reduce the amount of primer available to bind to the template DNA, leading to reduced amplification efficiency.
Hetero-dimer formation: Hetero-dimer formation occurs when two different primers anneal to each other, forming a double-stranded structure. This can happen when the primer sequences have complementary regions that allow them to hybridize to each other. Hetero-dimer formation can lead to non-specific amplification, as the primer pair may anneal to non-target DNA sequences in the sample.
Whats the optimal \( \Delta G \) for primer’s hairpin and self-dimer formations?
When designing primers for PCR, the goal is to minimize the formation of hairpins, self-dimers, and hetero-dimers, as they can lead to reduced amplification efficiency and non-specific amplification. One way to estimate the likelihood of these secondary structures forming is to calculate the Gibbs free energy (\( \Delta G \)) of the primer sequences.
A lower \( \Delta G \) value indicates a more stable structure, meaning that the secondary structure is more likely to form. Conversely, a higher \( \Delta G \) value indicates a less stable structure, meaning that the secondary structure is less likely to form. Therefore, the optimal \( \Delta G \) values for hairpin, self-dimer, and hetero-dimer formations are relatively higher values that are less negative (less stable), indicating that the primer sequences are less likely to form secondary structures.
The optimal \( \Delta G \) values for primer design can vary depending on the specific PCR conditions and the length and sequence of the primer. However, as a general rule of thumb, \( \Delta G \) values for hairpins and self-dimers should be higher than -9 kcal/mol and -6 kcal/mol, respectively. For hetero-dimers, the optimal \( \Delta G \) value can vary depending on the specific primer pair and PCR conditions, but typically \( \Delta G \) values below -5 kcal/mol are recommended to minimize non-specific amplification.
How To Design Primers
Tools needed:
- Primer3Plus (Terminal or Web)
- PCR Stat
- UCSC In Silico
- NCBI Genome Viewer (or ENSEMBLE Gene Viewer)
First, identify your amplification target, whether its a mutation point, or a gene. To identify mutation points (e.g., CREBBP.exon5:c.1318C>T), you can open NCBI genome viewer and search the whole HGVS nomenclature term from the Tools section.
Then select the region you want to design primer in, which depends on how long you want your product size to be, and then download the sequence in FASTA format.
After that, you can use primer3plus to pick primers with your optimal settings (GC, Tm, etc.).
To check the quality and eligibility of your primers, you can use PCR Stat web interface to see if they show any sign of dimerization. Finally, you can use UCSC In Silico web-tool to double check your PCR product.